


Kitzerow’s Autism and the Comorbidities Theory
“Autism and the comorbidities co-occur because they are caused by the same thing: gene mutations that trigger a system wide stress response, activating a BH4 Shunt. The body prioritizes this attempt to biochemically resolve the stress response over maintaining typical development and typical function in biochemically predictable ways. However, because the stress response is triggered by a gene mutation, it does not turn off, resulting in a lifelong allostatic existence.”
What is Kitzerow’s Autism and the Comorbidities Theory?
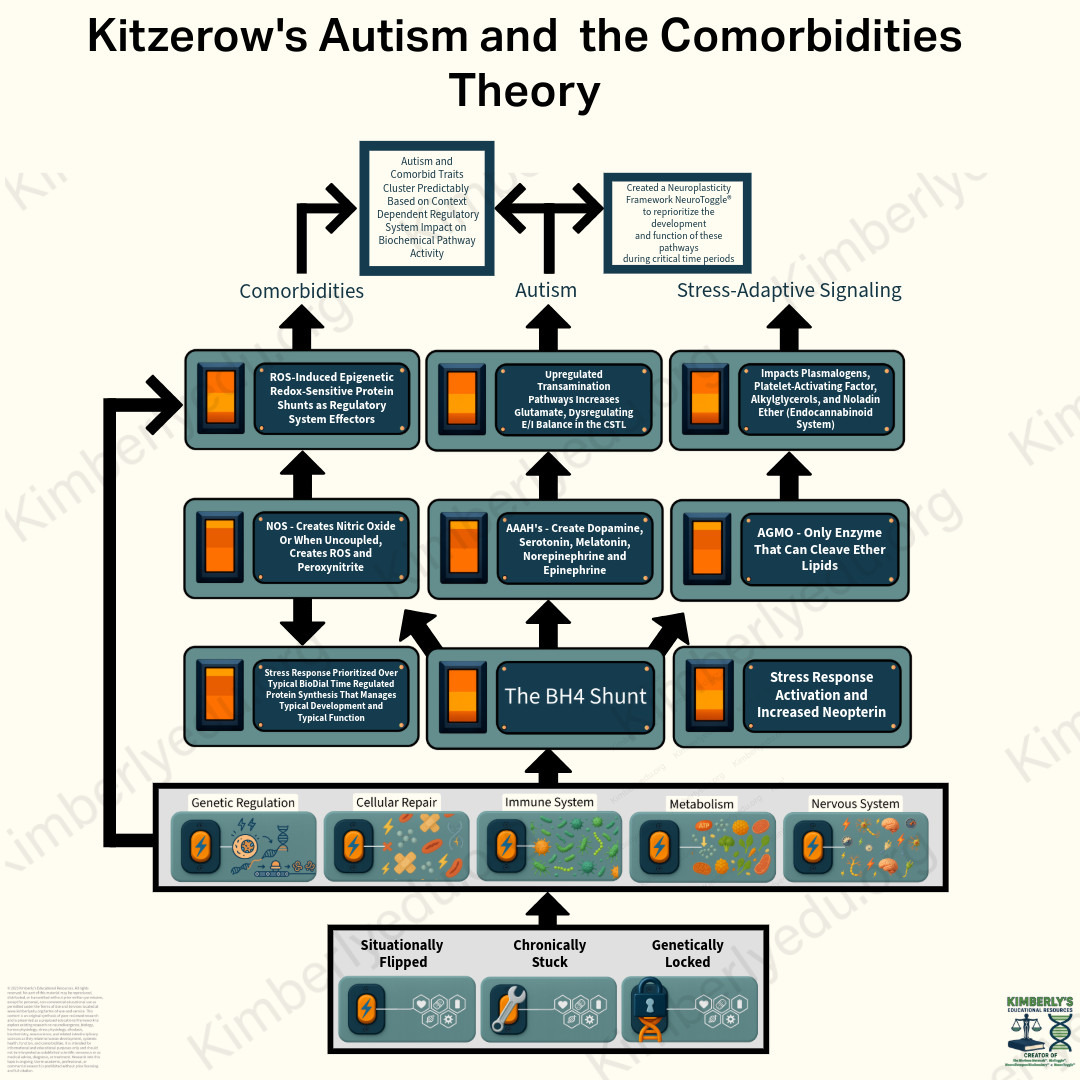
Genetic and epigenetic factors activate stress-response regulatory systems and trigger a BH4 Shunt, which simultaneously alters several BH4-dependent pathways. This includes dysregulated neurotransmitter synthesis through AAAH activity (reduced dopamine and serotonin output with increased glutamate via upregulated transamination pathways), changes in stress-adaptive signaling and endocannabinoid regulation through AGMO activity, and redox-driven biochemical pathway shifts through NOS uncoupling. These changes disrupt excitatory/inhibitory balance in the cortico-striatal-thalamic loop, producing autism traits. Over time, continued redox activation drives broader biochemical pathway shifts that contribute to systemic comorbidities. Regression may occur when excitotoxic stress exceeds neural tolerance. Distinct phenotypes emerge depending on which regulatory system or systems are genetically or epigenetically activated altering biochemical pathways in distinct ways as regulatory system effectors.
Brief Overview of Existing Literature That Supports This - More detail can be found in my ReseachGate Papers
Chronic allostasis, driven by epigenetic and genetic factors, initiates systemic changes in which and what forms of proteins are active, and the impact begins early and compounds over time, which may alter how the brain and body develop in childhood, function across the lifespan, and ultimately age (Hoffman et al., 2023; McEwen & Wingfield, 2003; Guidi et al., 2020; McEwen & Gianaros, 2014; Blair et al., 2014; Danese & MCEwen, 2012; Kallen et al., 2021). This cascade includes developmental alterations in neural and physical growth, immediate disruptions in systemic biochemical function, and symptomatic changes in neural circuitry (Arnsten, 2009; Mualem et al., 2024), which may become particularly impactful within the excitatory/inhibitory balance within the Cortico-Striatal-Thalamic Loop (CSTL), a network critical for motor planning, cognitive flexibility, emotional regulation, and executive function (Martel et al., 2022). These CSTL-specific disruptions contribute directly to many of the core behavioral characteristics associated with autism, and has been heavily linked to autism in the existing peer reviewed literature (Soghomonian, 2024; Abbot et al., 2018; Li & Pozzo-miller, 2020; Fuccillo, 2016; Di Martino et al., 2011).
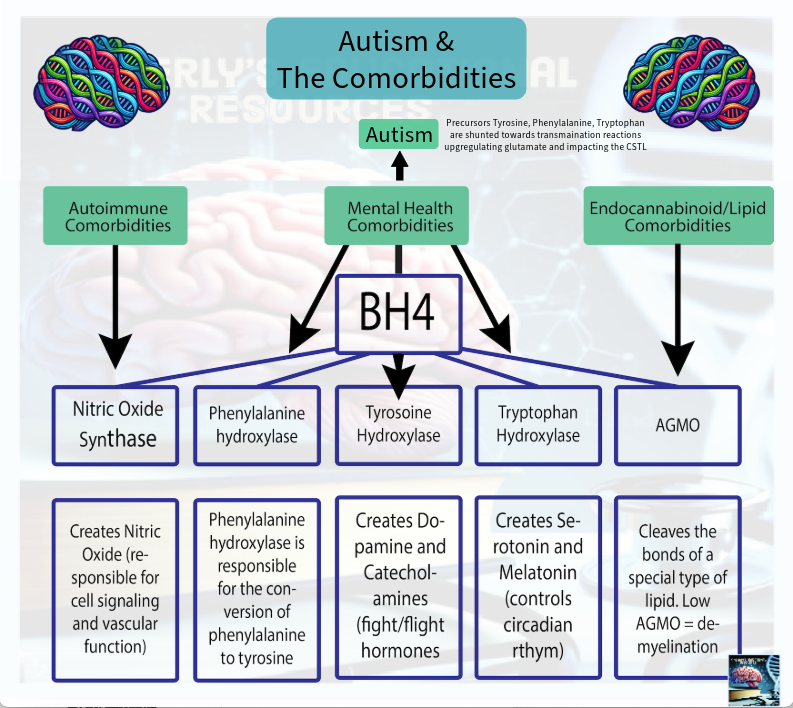
BH4 acts as the redox-sensitive biochemical lynchpin within this model, influencing each toggle: supporting or diverging nitric oxide synthase (NOS) in the immune system toggle; redirecting aromatic amino acid hydroxylases (AAAHs) in the nervous system toggle toward or away from dopamine, serotonin, and melatonin synthesis—shunting their precursor aromatic amino acids into transamination pathways; supporting alkylglycerol monooxygenase (AGMO) in the cellular repair toggle, as it is the only enzyme capable of cleaving ether lipid bonds; and contributing to metabolic adaptation via NOS uncoupling, which leads to reactive oxygen species (ROS) production. (Fanet et al., 2021).
The Four Core Pillars of Kitzerow’s 2023 Autism and the Comorbidities Theory and the 2025 Research that Supports This Model
Pillar 1: Genetic and epigenetic mutations activate internal stress-response systems.
Evidence:
• A 2025 study showed all diverse autism-associated mutations converge on a common cellular stress response.
• Research examining ASD-associated CNVs found shared activation of stress-adaptive signaling pathways across multiple genetic mutations.
Link: https://pmc.ncbi.nlm.nih.gov/articles/PMC12230234/
Pillar 2: Stress-response activation redirects biochemical pathway activity through the BH4 Shunt.
Evidence:
• Research shows GTPCH1, the rate-limiting enzyme of BH4 synthesis, is strongly induced by inflammatory and oxidative signaling.
• Studies report BH4 pathway disruption affecting monoaminergic and nitric-oxide dependent signaling in autism resulting in autism and comorbid traits.
Link: https://pubmed.ncbi.nlm.nih.gov/40002484/
Pillar 3: This disruption alters neurotransmitter synthesis and produces excitatory/inhibitory imbalance in the cortico-striatal-thalamic loop, contributing to autism traits.
Evidence:
• Stanford research identified reticular thalamus hyperexcitability within CSTL circuitry linked to autism behaviors and created a treatment that alleviated autism traits in all mice in the study.
Link: https://pmc.ncbi.nlm.nih.gov/articles/PMC12366697/
• Yale research examining glutamate receptors reports findings consistent with E/I imbalance and excitotoxic signaling.
Link: https://psychiatryonline.org/doi/10.1176/appi.ajp.20241084
Pillar 4: Biochemical pathway shifts contribute to systemic comorbidities and phenotypic clustering.
Evidence:
• Princeton research showed different categories of genetic mutations alter biochemical pathway activity that clusters autism and comorbid traits into different phenotypic and clinical outcomes.
Step 1: Eureka Moment - nonverbal autistic daughter can't blow out a candle on her birthday cake leads to the realization that autism and the comorbidities, including nonverbality, are physiologically linked rather than being independent traits.
Step 2: Hypothesis - It is biologically implausible for autism traits and comorbid traits to co-occur systematically in each phenotype without a shared biochemical root mechanism. This is now called the Exclusivity Principle.
Step 3: Testing Methodology - Created a biochemical network of gene-coded proteins and mapped autism-associated biomarkers onto the network to identify convergent points of regulatory dysregulation.
Step 4: Conclusion - The Autism and the Comorbidities Theory
- Core Mechanism: Autism arises from gene mutations and epigenetic factors that alter regulatory system behavior and constrain neural development and function. Comorbidities arise when stress-responsive BioToggle activation reallocates proteins via epigenetic redox-sensitive BH4 shunt trifurcation, producing predictable comorbidity clusters based on which regulatory system effectors are engaged. The timing and duration of BioToggle activation shape the comorbidity profile, with sustained activation increasing cumulative physiological impact via allostatic overload.
- Systems-Level Impact: BioToggles may be situationally triggered by environmental encounters, chronically activated when resolution fails, or genetically locked when mutations impair regulatory reset. Activation generates an allostatic response that is biochemically mediated through BH4-dependent regulation. Under cellular stress conditions GCH1 regulates BH4-dependent trifurcates of three epigenetic redox-sensitive shunts, producing predictable downstream effects across systems.
-
The BH4 Shunt and Autism Traits - (the AAAH's)
The Aromatic Amino Acid Hydroxylases are enzymes that are required for the creation the neurotransmitters dopamine, norepinephrine, epinephrine, serotonin, and melatonin.
BH4 is a required part of the recipe that combines with the AAAH’s to create these neurotransmitters.
Mechanism that Produces Autism Specific Traits: When BH4 is shunted, the precursor aromatic amino acids (phenylalanine, tryptophan, and tyrosine) are shunted towards transamination pathways. This alters glutamate activity. This change in glutamate activity/synthesis dysregulates the excitatory/inhibitory balance within the cortico striatal thalamic loop. The cortico striatal thalamic loop regulates movement, habit formation, and reward. This is what mechanistically alters neurodevelopment and behavior physiologically resulting in autism specific traits.
Validation: Stanford’s Neurodiversity Project reached out to me November 2023 asking for more information. Jan 2024 I petitioned for their help. August 2025 a team from Stanford published their version of a cure that targets this mechanism, E/I balance within a specific section of the cortico striatal thalamic loop called the reticular thalamic nucleus.
-
The BH4 Shunt and Comorbid Traits - (NOS Uncoupling)
Mechanism that Produces Comorbid Traits: The NOS shunt reflects redox-regulated, BH4-mediated control of nitric oxide synthase. The BH4 shunt modulates NOS coupling state, permitting regulated NOS uncoupling under stress conditions and shifting nitric oxide production toward reactive oxygen species generation. This redox shift selectively activates downstream epigenetic redox-sensitive protein shunts that function as regulatory system effectors.
Epigenetic Redox-Sensitive Protein Shunt Effectors Regulatory Architecture:
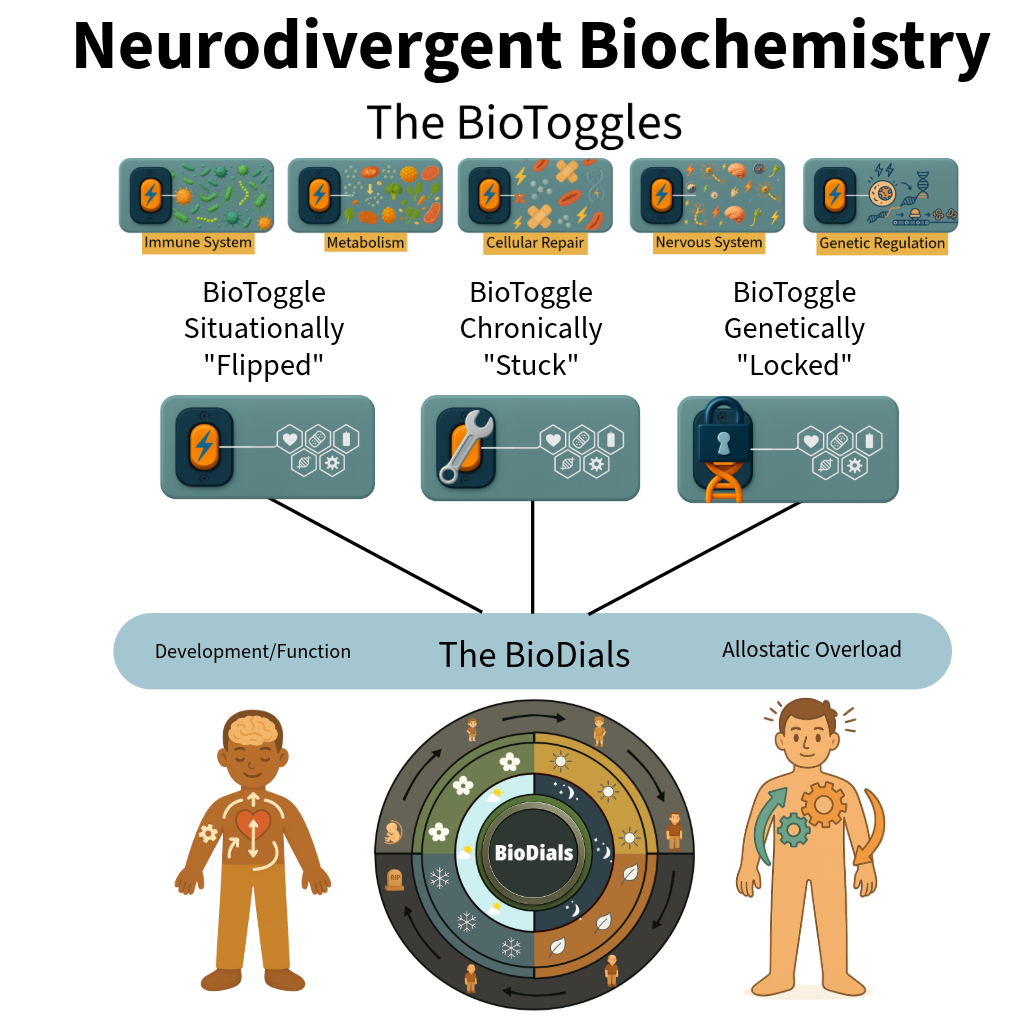
The BioToggles are five interdependent regulatory systems that respond to physiological stress through conserved, coordinated activation and context-dependent allostatic resource reallocation to support survival. Activation of the stress response disrupts the BioDials, prioritizing regulatory and survival demands over typical development and typical function in biochemically predictable ways.
The BioDials are four time-regulated protein synthesis mechanisms that maintain the body's steady kinetic flow of protein production across circadian, seasonal, developmental, and lifespan timescales. Preservation of this kinetic flow state is essential for continued physiological function and survival. Prolonged displacement of BioDial-regulated synthesis by stress-responsive BioToggle activity results in allostatic overload with biochemically predictable consequences, the comorbidities.
Individual Variation: Individual differences arise from two mechanisms: experience-driven neural development, which is primarily shaped postnatally by environmental input, and regulatory system activation, in which genetically and epigenetically constrained BioToggles are situationally activated beyond the genetic phenotype. The degree, duration, and timing of this BioToggle activation disrupt BioDial-regulated protein synthesis, reallocating resources toward survival and producing variation in development and function.
Timing Effects: Trait severity and presentation depend on when a gene mutation is functionally relevant, which regulatory system it affects, when BioToggles are activated relative to BioDial-regulated protein synthesis, and how experience shapes neural development and function.
BioToggle activation during sensitive developmental windows more strongly displaces developmental and maintenance processes, while experience-driven neural plasticity during these periods further refines or amplifies functional outcomes.
-

The BH4 Shunt and the Endocannabinoid System - (AGMO)
Mechanism: AGMO is a BH4 dependent enzyme. It is the only enzyme known to cleave the bond of ether lipids in humans. One such ether lipid is noladin ether, which creates 2-AG one of the most abundant endocannabinoids.
Other examples of ether lipids include plasmalogens, alkylglycerols, and platelet-activating factor (PAF).
The AGMO shunt impacts ether lipid cleavage required for endocannabinoid system activity, cellular repair activity, and detoxification. Disruption of noladin ether cleavage to 2-AG alters endocannabinoid signaling. Prolonged AGMO impairment contributes to lysosomal dysfunction, reduced repair capacity, and cumulative system wear.