


Kitzerow’s Autism and the Comorbidities Cascade
“Gene mutations and epigenetic factors trigger a stress response that is prioritized over typical development and function, resulting in biochemically predictable autism and comorbid traits.” - Kimberly Kitzerow
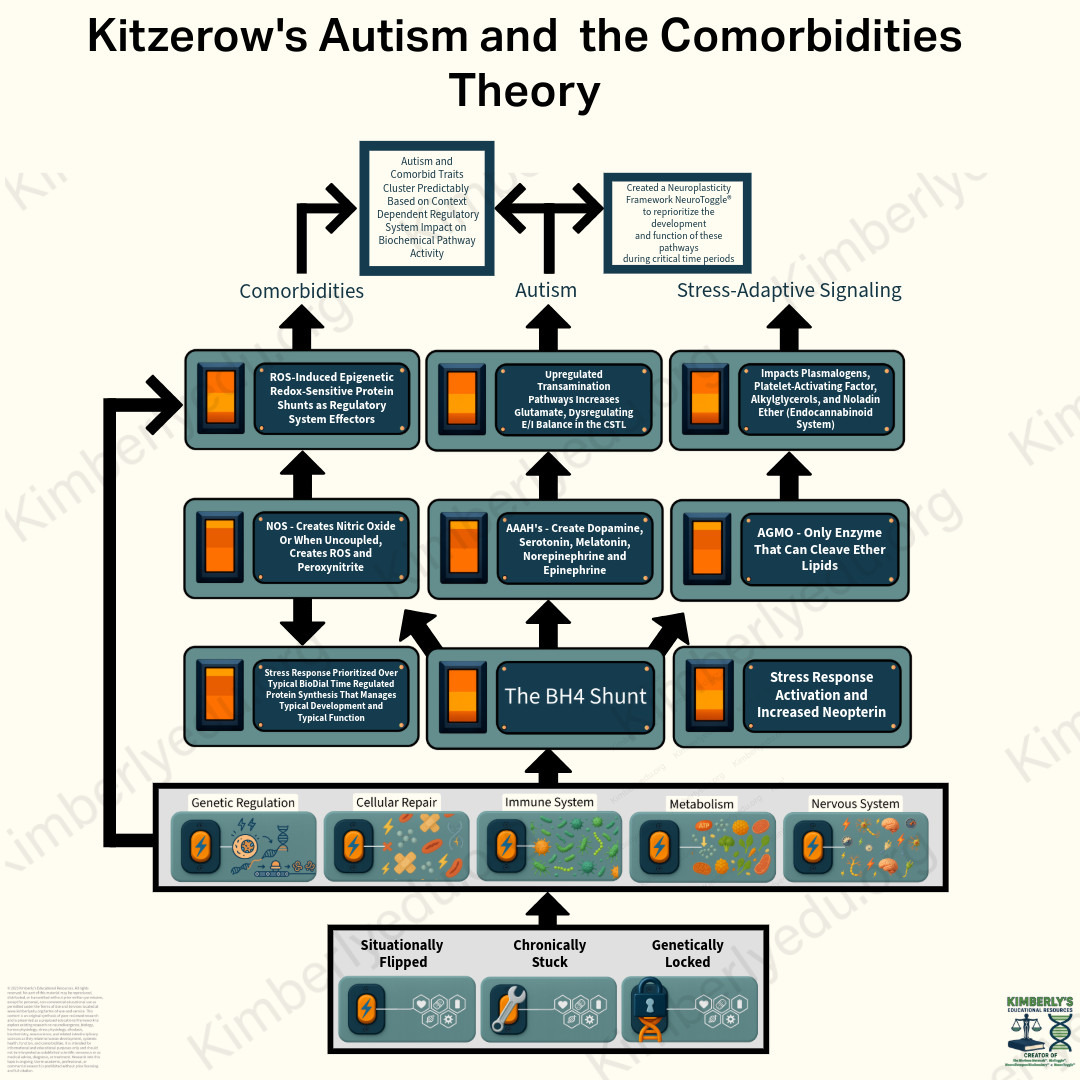
Kitzerow’s Autism and the Comorbidities Cascade
A systems-level theoretical model for understanding how different genetic and epigenetic inputs may converge on a shared biochemical cascade that affects neural development and systemic function.
What’s Happening Biologically?
Genetic and epigenetic factors can keep the body’s stress-response system activated during neural development.
Within the model, biological resources are reallocated through the BH4 Shunt toward survival rather than typical neural-circuit development and systemic function.
The impact on neural development leads to autism traits, while the impact on systemic function leads to comorbid traits.
Why Do Different Phenotypes Develop?
Different patterns of impact across biologically interconnected pathways, occurring during different key developmental and biological periods, lead autism traits and comorbid traits to cluster into different phenotypes.
Where the Model Stands Now
The model remains theoretical while evidence is evaluated across its individual mechanisms and its larger systems-level architecture.
Current Progress
Tracking movement from theoretical and emerging evidence toward converging evidence, diagnostic development, and treatment development.
Why Autism Traits and Comorbid Traits Share the Same Mechanism
Autism traits and common comorbid traits are not presented as separate biological events in this model. They are two downstream expressions of the same upstream stress-response cascade.
The Shared Starting Point
Different genetic and epigenetic inputs can keep the stress-response system activated. The proposed BH4 Shunt then reallocates biological resources toward survival priorities.
From that shared mechanism, the cascade branches according to where the biological impact is expressed.
Autism Traits
When resource reprioritization affects neural-circuit development, it changes how circuits supporting communication, movement, learning, sensory processing, regulation, behavior, and adaptive skills develop.
Comorbid Traits
When the same cascade affects function, development, and accumulated load across interconnected regulatory systems, comorbid traits emerge across immune, metabolic, cellular-repair, nervous-system, and genetic-regulation domains.
How the Cascade Leads to Autism Traits
Neural circuits store the information needed to perform skills and behaviors. When the developmental environment changes, different circuits can develop differently, producing the strengths, challenges, and diagnostic traits associated with autism.
Because individual circuits are affected differently, autistic people may present at opposite ends of the same skill or behavior spectrum.

Autism Trait Pathways
| BH4 Dependent Pathways | Primary Function |
|---|---|
| BH4 Shunt | Central regulatory pathway underlying the autism trait cascade |
| AAAH Shunt | Catecholamine regulation and excitatory-inhibitory balance in neural circuits controlling movement, habit formation, reward, communication, and sensory processing |
| AGMO Shunt | Endocannabinoid signaling, stress signaling, lipid signaling, and neurodevelopment |
| NOS Shunt | Nitric oxide signaling, oxidative stress, mitochondrial function, synaptic function, and nervous system regulation |
How Different Neural-Circuit Effects Can Present
| Skill or Behavior | Possible Presentations |
|---|---|
| Reading | Hyperlexia or dyslexia |
| Communication | Hyperverbal or nonverbal |
| Sensory Processing | Sensory seeking or sensory avoiding |
| Learning | Savant-like skills or difficulty with daily living skills |
| Behavior | Impulsive or rigid |
| Interests | Broad interests or highly focused interests |
| Adapting to Change | Novelty seeking or resistance to change |
| Social Interaction | Highly social or socially withdrawn |
| Memory | Exceptional memory or difficulty with memory and recall |
NeuroToggle® addresses skills and behaviors through the neural circuitry that produces them, using neuroplasticity-informed teaching experiences to build, strengthen, expand, and time neural circuits.
How the Cascade Leads to Comorbid Traits
The same upstream stress-response cascade can simultaneously affect biologically interconnected systems throughout the body.
More than 95% of autistic individuals also experience at least one comorbidity. These can include ADHD, gastrointestinal disorders, chronic pain, anxiety, connective tissue disorders, dysautonomia, immune differences, sleep disorders, and many other co-occurring conditions.
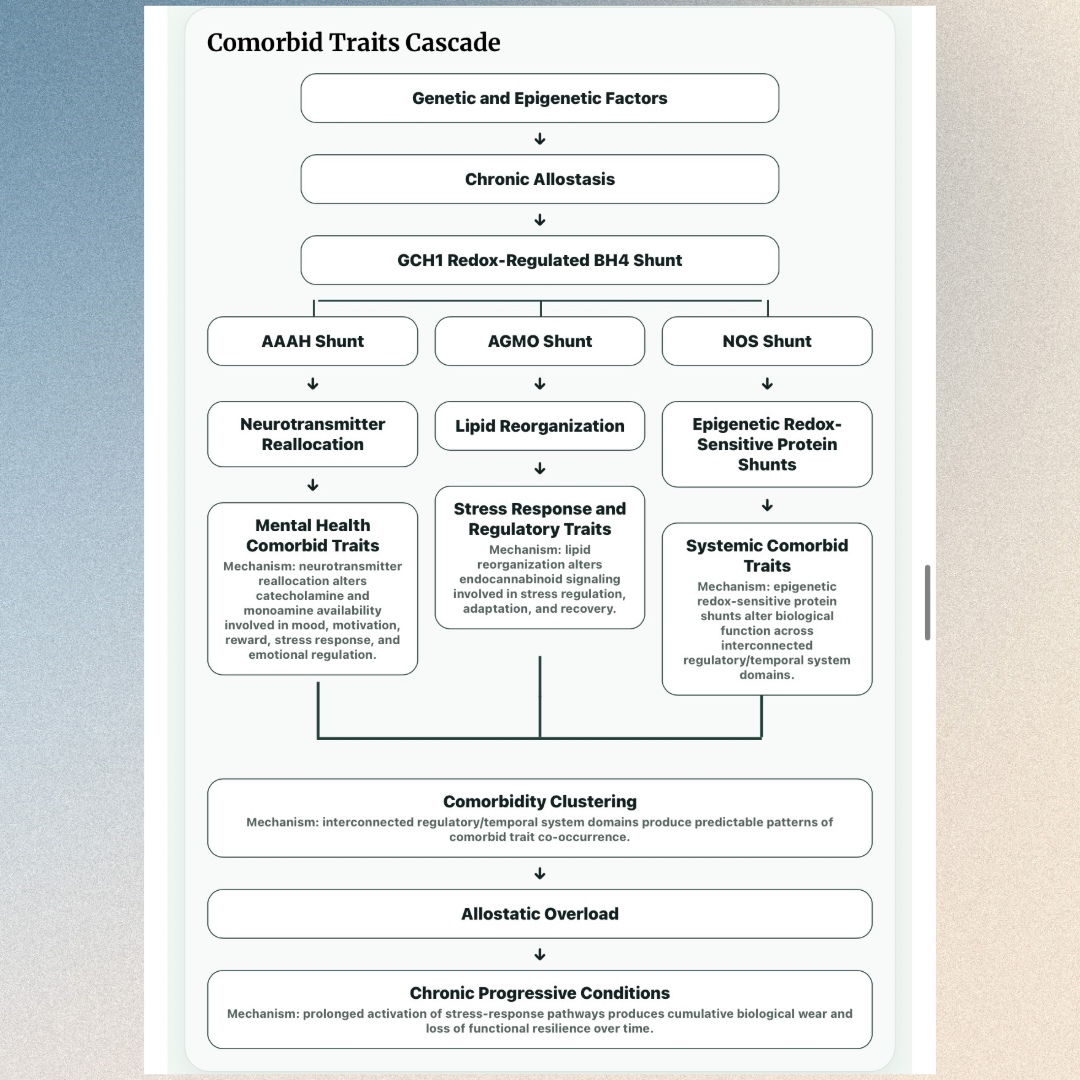
Comorbid Trait Pathways
| Regulatory System Domain Activation | May Influence |
|---|---|
| Immune Regulation | Immune-related traits and inflammatory responses |
| Metabolic Regulation | Energy production, metabolism, gastrointestinal function, and fatigue-related traits |
| Cellular Repair | Connective tissue, extracellular matrix remodeling, healing, and tissue integrity |
| Nervous System Regulation | Autonomic function, pain regulation, sleep, migraine, and dysautonomia-related traits |
| Genetic Regulation | Epigenetic adaptation, gene expression, and long-term biological regulation |
BioToggle® explains how effects across interconnected regulatory domains contribute to recurring comorbid trait clusters.
How Systemic Effects Appear Across Regulatory Domains
Use the tabs to see what each domain regulates, what its effects can look like, and why it matters.
Immune System Differences Can Show Up Alongside Autism
What It Regulates
Inflammation, immune response, illness signaling, and how the body reacts to internal and external stressors.
What It Can Look Like
Autoimmune patterns, autoinflammatory responses, frequent illness, strong inflammatory responses, or broader immune dysregulation.
Why It Matters
Persistent immune activation can affect regulation across the body and contribute to broader physiological stress patterns.
Metabolic Differences Can Shape Daily Function
What It Regulates
Digestion, nutrient use, energy production, metabolic balance, and how the body fuels development and function.
What It Can Look Like
GI problems, food sensitivities, unstable energy, feeding issues, metabolic differences, or obesity.
Why It Matters
When metabolism is strained, effects can appear in daily function and broader physical regulation.
Cellular Repair Differences Affect Structure and Recovery
What It Regulates
Connective tissue integrity, structural support, tissue maintenance, and physical repair processes.
What It Can Look Like
Joint instability, slow recovery, chronic pain, tissue fragility, or altered pain perception.
Why It Matters
Structural and repair differences can shape physical stability, comfort, and how biological stress is carried through the body.
Nervous System Differences Affect Regulation and Stability
What It Regulates
Stress response, autonomic function, emotional processing, neurological stability, and internal-state regulation.
What It Can Look Like
PoTS, anxiety, OCD, ADHD, seizures, tics, FND, burnout, dysregulation, or unstable nervous-system states.
Why It Matters
Nervous-system dysregulation can affect cognition, emotion, movement, autonomic function, and daily regulation simultaneously.
Genetic Regulation Shapes Biological Timing and Adaptation
What It Regulates
Gene expression, epigenetic adaptation, protein prioritization, biological timing, and long-term regulatory response.
What It Can Look Like
Sleep and circadian disruption, irregular biological rhythms, and longer-term changes in regulatory patterns.
Why It Matters
Changes in genetic regulation can influence when biological processes occur and how other regulatory systems adapt over time.
Why Different Autism and Comorbid Trait Clusters Develop
A phenotype is the observable cluster of autism traits and comorbid traits expressed by an individual. In this model, the cluster reflects which systems are affected, how long activation persists, and when the impact occurs.
Which Regulatory Domains Are Affected?
Immune regulation, metabolic regulation, cellular repair, nervous-system regulation, and genetic regulation are interconnected. The affected domains determine which pathways, functions, and resources are involved.
How Long Does Activation Persist?
Situational, chronically stuck, and genetically locked activation determine the persistence and extent of the biological impact.
When Does the Impact Occur?
Ultradian, circadian, circannual, developmental, and age-related timing determine whether the effect is expressed through current function, development, or accumulated wear and load.
Temporal System Domains
| Temporal System Domain | What It Describes | Why Timing Matters |
|---|---|---|
| Ultradian | Short cycles that repeat multiple times within a day | Shapes rapid, repeating changes in biological activity |
| Circadian | Daily cycles organized across approximately 24 hours | Influences sleep, hormones, metabolism, immune activity, and nervous-system regulation |
| Circannual | Seasonal and yearly biological cycles | Influences longer-term patterns in regulation and function |
| Developmental | Time-sensitive stages when systems form, mature, and specialize | Impact during neural development can change how circuits underlying skills and behaviors develop |
| Age Cycles | Age-related changes in capacity, repair, adaptation, and resilience | Changes how the same regulatory demand may affect a person across the lifespan |
BioToggle × Activation Duration
| BioToggle | Situational Activation | Chronic Activation | Genetically Locked Activation |
|---|---|---|---|
| Immune | Immune Trigger | Immune Overload | Immune Lock |
| Metabolic | Metabolic Shift | Metabolic Overload | Metabolic Lock |
| Cellular Repair | Repair Trigger | Repair Overload | Repair Lock |
| Nervous System | Nervous Activation | Nervous Overload | Nervous Lock |
| Genetic Regulation | Protein Demand | Protein Overload | Protein Lock |
How to Read the Phenotype Map
Each phenotype represents a BioToggle × duration × BioDial combination. Different combinations create different effects on neural development and systemic function. This is why autistic individuals can share core diagnostic traits while having different strengths, challenges, comorbidities, and developmental trajectories.
Kitzerow’s Autism and the Comorbidities Cascade
This cascade shows how a stimulus creates regulatory system activation, how a BioToggle detects setpoint deviation, how an allostatic state is induced, how the BH4 Shunt reallocates resources, and how BioToggle × BioDial impact produces trait outcomes over time.
Stimulus occurs
A stimulus may be genetic, chronic, or situational. This determines whether activation is built into the system, fails to resolve, or resolves after a temporary trigger.
A BioToggle detects setpoint deviation
The stimulus creates deviation inside a regulatory system domain. These domains are the BioToggles in Kitzerow’s model.
The regulatory loop activates response
Each BioToggle has its own domain-specific version of the regulatory loop. The roles are shared, but the biological components are unique to each regulatory system domain.
These are descriptive roles, not one identical biological structure. Each BioToggle has domain-specific sensors, setpoints, controllers, and effectors.
An allostatic state is induced within the affected BioToggle
The affected regulatory domain shifts into an allostatic state to resolve the source of stress. The body prioritizes regulatory system effectors needed for stress resolution and survival.
The BH4 Shunt reallocates biological resources
Under allostatic demand, the BH4 Shunt reallocates biological resources toward survival-focused regulatory effectors and away from typical BioDial activity.
Typical BioDial activity is deprioritized
BioDials represent the ongoing flow of protein synthesis across time. Under allostatic activation, timing-based protein synthesis is deprioritized while stress-resolution effectors are prioritized.
BioDial impact alters function, development, and wear/load
Function, development, and wear/load are BioDial impact patterns that shape how each BioToggle domain is affected over time.
Trait outcomes emerge through BioToggle × BioDial impact
Trait outcomes reflect how each regulatory system domain is affected through BioDial impact patterns of function, development, and wear/load.
Extent of impact depends on timing and duration
Core autism traits reflect nervous system developmental impact, where neural circuit development affects skills and behaviors. Comorbid traits reflect altered function, development, and wear/load across other regulatory system domains.
What can be done about it?
Because autism traits and comorbid traits reflect different expressions of the cascade, the support pathway depends on what is being targeted.
Autism traits and comorbid traits reflect how a multivariable stress-response system cascade is expressed across regulatory system domains and biological timing systems.
How This Theoretical Model Was Built
Kitzerow’s Autism and the Comorbidities Theoretical Model was developed through the Jigsaw Puzzle Research Methodology, a computational systems analysis approach that builds a conserved species-level biochemical reference framework first, then compares demographic-level biomarker findings against that framework to identify recurring points of dysregulation and reconstruct a coherent biochemical cascade.
Rather than treating biomarkers, pathways, and studies as isolated findings, this methodology treats them as pieces of a larger biological structure that must be assembled into a coherent functional model.
A biochemical network of gene-coded proteins is constructed to define the conserved species-level functional blueprint used as the reference system.
Demographic-level biomarker datasets are mapped onto that reference framework so population-level variation can be compared against shared biological architecture.
Recurrent deviations from the conserved framework are identified as consistent points of dysregulation across datasets, pathways, and regulatory systems.
Those recurring patterns are traced across pathways to reconstruct the biochemical cascade linking autism traits and comorbidities to shared system-level dysregulation.
In this model, the repeated co-occurrence of autism traits and comorbidities is treated as a structured biological pattern rather than a random collection of separate findings. The methodology was used to test whether those repeated outcomes could be explained through one coherent biochemical mechanism.
Kimberly’s ResearchGate Articles and Converging Evidence
Kimberly’s articles document the proposed mechanisms, pathway relationships, and predictions of the model so they can be independently evaluated against research published before and after the model was introduced.
ResearchGate Articles
Kimberly’s ResearchGate articles document the prior research literature used to trace the BH4, AAAH, NOS, and AGMO pathway mechanisms; regulatory-system effects; neural-circuit implications; and the proposed autism and comorbidities cascade.
Track Research Progress
Follow movement from a theoretical model through emerging and converging evidence toward diagnostic and treatment development.
Later Cascade-Level and Phenotype Convergence
Later publications reproduce substantial portions of the ordered cascade and phenotype architecture Kimberly had already publicly documented. Kimberly’s comparison reports evaluate both the structural convergence and whether the later work reflects independent derivation or use of an already disseminated framework.
UCSD–Naviaux Three-Hit Model
Kimberly’s report finds that Naviaux’s earlier Cell Danger Response model remained centered on mitochondrial signaling, metabolic shift, and chronic disease physiology. The 2025 three-hit expansion reorganized it into the same directional progression already present in Kitzerow’s cascade: three stress categories → metabolic shift → E/I dysregulation → autism and comorbidities → developmental timing → neuroplasticity relevance.
The report characterizes this as full cascade replication with confirmed affiliation, because the overlap is in the ordered architecture rather than isolated mechanisms.
Melillo vs. Kitzerow
Kimberly’s report finds that Melillo’s later developmental neuroimmune cascade follows the same top-to-bottom direction: immune dysregulation → E/I imbalance → synaptic-pruning dysregulation → altered connectivity and developmental outcomes → neuroplasticity.
The key difference is the middle of the cascade. Kitzerow’s earlier model identifies the BH4 Shunt and its AAAH, NOS, and AGMO branches as the biochemical mechanisms connecting immune-domain activation to circuitry, pruning, connectivity, and social-behavioral outcomes; Melillo’s later cascade moves directly from immune mechanisms to those downstream neural effects.
Princeton Phenotype Study
Kimberly’s report finds that Princeton tested the same causal architecture she had previously documented: categories of gene mutations → distinct biological pathway changes → predictable clusters of autism traits and comorbidities.
The comparison distinguishes the methods. Kitzerow built a species-level network of gene-coded proteins and mapped autism biomarkers against it to identify biologically constrained dysregulation; Princeton used existing SPARK data and bioinformatics to group trait combinations and connect the resulting subtypes with genetic profiles. The report notes that Kitzerow’s public articulation predates Princeton’s publication by approximately 26 months.
What the Comparisons Establish
These later publications provide substantial cascade-level and phenotype-level convergence. Kimberly’s reports argue that the similarity extends beyond shared topics to ordered structure, causal sequence, and predicted clustering, while direct experimental testing is still needed for the complete BH4-centered biochemical middle and its specific BioToggle × BioDial relationships.
Redox-Sensitive GCH1/BH4 Resource Reallocation
A 2025 systematic review of tetrahydrobiopterin in autism describes the same central biochemical pattern proposed in Kimberly’s model: inflammatory and oxidant signals regulate GCH1, BH4 metabolism shifts, neurotransmitter-related BH4 availability falls, and BH4 may be diverted toward antioxidant or nitric-oxide-related inflammatory activity.
BH4 Pathway Shift as a Central Autism Mechanism
Kimberly identifies this review as direct convergence with the proposed redox-sensitive GCH1/BH4 Shunt: biological demand reallocates BH4-dependent resources away from typical monoaminergic neurotransmission and toward survival-related redox, antioxidant, inflammatory, and nitric-oxide functions.
The review reports altered BH4 metabolism in autistic individuals, including lower BH4, altered neopterin, higher nitric oxide, and a proposed shift in BH4’s metabolic role. Within Kitzerow’s cascade, this is the biochemical middle connecting stress-response activation with neural-development effects that produce autism traits and systemic effects that contribute to comorbid traits.
Mechanism-Targeted Approaches in Experimental Models
These studies move beyond association by experimentally targeting downstream circuit or receptor dysfunction predicted within the model. They are preclinical findings, not established treatments for autistic people.
Reticular Thalamic Hyperexcitability and Z944
Jang and colleagues found that hyperexcitability of the reticular thalamic nucleus contributed to autism-related behaviors in a Cntnap2 mouse model. Pharmacological suppression with the T-type calcium-channel blocker Z944 and chemogenetic inhibition of reticular-thalamic activity significantly improved social deficits, repetitive behaviors, hyperactivity, and seizure-related phenotypes.
Kimberly’s comparison identifies the reticular thalamus as a specific node within the cortico-striatal-thalamic loop and interprets the study as treatment-level support for the functional role of excitatory–inhibitory imbalance within the downstream autism-trait cascade.
SLC6A20 Inhibition and NMDAR Rescue
Roh and colleagues found that antisense inhibition of the glycine transporter Slc6a20a restored NMDA-receptor function, rescued abnormal synaptic phospho-proteomic signaling, and normalized autism-related phenotypes in Shank2- and Shank3-mutant mice, with model-dependent rescue profiles.
Targeting human SLC6A20 also restored suppressed NMDAR function in cortical organoids carrying SHANK2 or SHANK3 mutations. The findings support the possibility of matching a mechanism-targeted approach to a defined form of receptor hypofunction while also showing that responses can differ by genetic model.
Important: These approaches were tested in mouse models and, for the SLC6A20 study, human cortical organoids. They identify treatment-relevant mechanisms for further research but do not establish safety, effectiveness, dosing, or clinical suitability for autistic children or adults.
Where to Start
Choose the path that matches what you are looking for.
Review the Evidence
Full framework, methodology, validation, and supporting data.
Start Here → ParentsSupport Your Child
Understand development, learning, and practical application.
Start Here → EducatorsTransform Instruction
Apply neuroplasticity-based teaching to build skills and behavior.
Start Here → NeurodivergentsNeurodivergent Biochemistry
See how the model explains development, function, and experience.
Start Here →