


Statement of Summary & Conceptual Framework
Statement of Summary & Conceptual Framework
By Kimberly Kitzerow, B.S.
Title: Neurodivergent Biochemistry and the Autism and the Comorbidities Theory
What is The Autism and the Comorbidities Theory?
In May 2023, I first introduced the foundation of this hypothesis in a TikTok video, proposing that autism traits and associated medical comorbidities stem from a unified biochemical disruption: the dysregulation of tetrahydrobiopterin (BH4). Over the following months, I expanded this concept by integrating linked comorbidities and stress-response mechanisms, gradually constructing a systems-level framework that accounts for both neurodevelopmental outcomes and the chronic health conditions commonly observed in neurodivergent individuals—and how these evolve across the lifespan.
Autism and the Comorbidities Theory: My hypothesis (now turned theory) describes the pathology underlying autism and its comorbidities as arising from gene mutations that lock the body into a persistent state of allostatic stress-regulation mode. This genetically locked stress causes epigenetic changes that alter which pathways and proteins are active, directing development and function toward long-term stress adaptation rather than typical development and function. Specifically, the BH4 Shunt results in the physiological traits of autism, and the other simultaneous epigenetic redox-sensitive shunts are responsible for the comorbidities, biochemically linking them. Over time, this leads to atypical development, functional differences, and allostatic overload, producing the neurodivergence and comorbid conditions associated with autism.
Chronic allostasis, driven by epigenetic and genetic factors, initiates systemic changes in which and what forms of proteins are active, and the impact begins early and compounds over time, which may alter how the brain and body develop in childhood, function across the lifespan, and ultimately age (Hoffman et al., 2023; McEwen & Wingfield, 2003; Guidi et al., 2020; McEwen & Gianaros, 2014; Blair et al., 2014; Danese & MCEwen, 2012; Kallen et al., 2021). This cascade includes developmental alterations in neural and physical growth, immediate disruptions in systemic biochemical function, and symptomatic changes in neural circuitry (Arnsten, 2009; Mualem et al., 2024), which may become particularly impactful within the excitatory/inhibitory balance within the Cortico-Striatal-Thalamic Loop (CSTL), a network critical for motor planning, cognitive flexibility, emotional regulation, and executive function (Martel et al., 2022). These CSTL-specific disruptions contribute directly to many of the core behavioral characteristics associated with autism, and has been heavily linked to autism in the existing peer reviewed literature (Soghomonian, 2024; Abbot et al., 2018; Li & Pozzo-miller, 2020; Fuccillo, 2016; Di Martino et al., 2011).
If you have concerns about tetrahydrobiopterin (BH4) deficiencies, you can share the Consensus Guidelines for the Diagnosis and Treatment of Tetrahydrobiopterin (BH4) Deficiencies with your doctor. These guidelines provide helpful information for healthcare professionals. Always defer to medical guidance from your doctor or healthcare provider for any decisions regarding diagnosis or treatment. You can access the current guidelines here.

What is Neurodivergent Biochemistry?
Neurodivergent Biochemistry looks at how the body's survival systems, genetics, and environment work together to shape development, health, and learning in all forms of neurodivergence. At its core are the BioToggles™ (survival switches) and BioDials™ (timing systems) that control which proteins the body makes and when. When a BioToggle is switched on, the body changes its priorities, focusing on protection instead of growth. This creates predictable patterns in traits, behavior, and health often seen in neurodivergents.
Central Dogma of Molecular Biology Expanded
Each gene holds the instructions for making one protein (and different isoforms, or "flavors," of it) that are "made to order" for the conditions needed.
What are Proteins?
Think of the body's functional blueprint, its gene-coded biochemical network, as a complex conveyor belt system. Each protein is a worker carrying out a specific job. Every single function in the body is managed by one of these workers.
BioDials and the Kinetic Flow-State (Dynamic Equilibrium)
Typically, there is a steady kinetic flow of proteins being created to keep the body typically developing and typically functioning.
This flow state is regulated by the BioDials through daily, seasonal, developmental, and age-related timing rhythms.
Stress and Epigenetics
Cellular stress interrupts this flow, flipping BioToggles and activating "turbo mode" to restore baseline. This adaptive state is called allostasis. In allostasis, proteins needed for the stress response are turned on, I call these allostatic proteins. While the creation of other proteins are shut down to conserve resources and protect certain vital functions. This process (epigenetics) helps us survive by shifting into "emergency mode." | call these shifts Epigenetic Redox-Sensitive Shunts.
Neurodivergent Biochemistry and An Allostatic Existence
Sometimes the stress switches (BioToggles) don't flip back after the stress passes. Instead of returning to baseline, they remain engaged. This keeps the body stuck in allostasis, constantly trying to restore balance but never fully reaching it. This disrupts the body's natural flow of protein synthesis. When Biologgles stay locked, the BioDials that normally regulate typical development and typical function (day-night cycles, developmental stages, and age-related rhythms) can't run properly. The result is a chronic diversion of resources: proteins that should support typical development and typical function are suppressed, while stress-related proteins stay prioritized. Over time, this imbalance drains energy, creates wear on multiple systems, and alters both development and function in predictable biochemical patterns. I call this Neurodivergent Biochemistry.
Neurodivergent Biochemistry Framework: Delineates and organizes data on how the body's stress-response regulatory systems (BioToggles) and timing-based protein synthesis mechanisms that sustain the biochemical flow-state of life (BioDials) operate in a feedback loop as part of the allostatic survival response.
When stress disrupts the flow-state, BioToggles flip and activate effectors such as epigenetic redox-sensitive shunts, which epigenetically change which proteins are turned on and off to participate in survival mechanisms that work to restore baseline and protect those that need to be conserved.
This process exists to keep us alive during situational BioToggle flips, but when BioToggles become stuck or genetically locked, the shunts remain persistently engaged, diverting resources away from typical BioDial function toward survival turbo-mode mechanisms.
This long term shift produces the predictable biochemical patterns within epigenetic redox-sensitive protein shunts, underlying neurodivergence and its comorbid conditions, as mapped through the BioGene Network:
These changes lead to:
Atypical development
Atypical function
Wear and tear over time
Why Is There So Much Variation in Phenotypes?
Neurodivergence can be understood as a prolonged state of allostasis, a continuous effort to maintain internal stability under stress that gradually alters physiological function and development.
My framework identifies five categories of regulatory systems, or BioToggles, that maintain biochemical balance within the body: the immune system, metabolism, cellular repair, the nervous system, and genetic regulation.
Each BioToggle monitors and restores its own baseline while interacting with the others through feedback loops. When one system is stressed, resources are reallocated across these networks through epigenetic redox-sensitive protein shunts, which act as biochemical effectors to restore equilibrium.
Phenotypic variation depends on several key variables:
Which BioToggle is active
Immune system – protects against pathogens (e.g., viruses, bacteria)
Cellular repair – responds to physical trauma (e.g., wounds, tissue damage)
Metabolism – prevents energy or nutrient failure (e.g., diabetic coma, starvation)
Nervous system – preserves neurological integrity (e.g., stroke, seizure)
Genetic regulation – maintains genomic stability (e.g., replication errors, epigenetic dysregulation)
The duration of activation
Situationally flipped: Temporarily activated in response to an environmental or biochemical stressor and capable of returning to baseline once resolved. Acute and resolves.
Chronically stuck: Persistently active due to prolonged stress exposure or disrupted feedback regulation due to gene mutations in allostatic proteins, or overloaded system, leading to sustained biochemical imbalance. Stuck and results in allostatic overload over time.
Genetically locked: Permanently altered through genetic mutations that impact the regulatory system resulting in set point deviations or ineffective regulatory system function. Lifelong and does not resolve.
The developmental stage during which activation occurs
Which BioDial processes (time-regulated protein synthesis) are affected
Circadian rhythm: Daily regulation of protein synthesis that maintains physiological balance in the body
Circannual cycles: Seasonal regulation of protein synthesis that coordinates metabolism, immunity, and repair with environmental and energetic demands during the change of seasons
Development: Time-regulated progression from childhood through adulthood
Aging and repair: Time-regulated cell turnover that protects barriers and repairs the body as part of natural processes
The specific biochemical pathway that is disrupted
The downstream physiological impact of the dysregulation
Together, these factors explain why individuals with similar genetic or diagnostic profiles may exhibit different traits, severities, or comorbidities within the neurodivergent spectrum. This is a systems biology explanatory framework: Neurodivergence isn't genetically homogeneous but mechanistically unified.

My Process
Autistic Biomarkers Overlapped with BH4 Pathway Shunt – First Pattern Identified
Following the public documentation of my daughter’s progress, specifically her achievement of functional speech—I was frequently asked how her nonverbal state related to her autism diagnosis. To explore this question, I began visually mapping potential connections using Adobe Illustrator, arranging peer-reviewed data into a jigsaw puzzle format to identify cross-disciplinary patterns.
This method allowed me to systematically examine overlapping biochemical, neurological, and immunological mechanisms. Through this process, I observed that biomarker data consistently pointed to dysregulation within a specific biochemical pathway. More precisely, the data revealed that this pathway was not simply underactive or overactive, but shunted—rerouted in a way that reflected a physiological shift in priorities under chronic stress.
The recurring presence of both autism traits and systemic comorbidities (At rates from 50-80% prevalence per comorbid condition, and more than 95% of autistics having at least one of them) in a specific subset of individuals suggested a unified, simultaneous genetic root cause. Comorbidities include sleep disorders, GI problems, metabolism disorders, neuroinflammation, altered inflammatory responses, immune abnormalities, anxiety and other mental health conditions, neurological disorders, lysomal storage disorders, seizures, tics, OCD, ADHD, motor delay, motor impairment, obesity, deafness, hearing loss, Functional Neurological Disorder (FND), Hypermobile Ehlers-Danlos syndrome (hE-DS), postural tachycardia syndrome (PoTS) and learning differences (Al-Betagi, 2021; Chung & Kim, 2024; Khachadourian et al., 2023; Bougeard et al., 2021; Burns et al., 2023; Hours et al., 2022; González-Herrero et al., 2022; Owens et al., 2021). This led to the identification of converging disruptions centered on the tetrahydrobiopterin (BH4) pathway—a pattern I would later define as the BH4 shunt.
I soon observed that this shift may also involved redox-sensitive proteins, which are proteins that react to reactive oxygen species, etc. to restore redox homeostasis. Redox homeostasis is maintenance of the balance between antioxidants vs antioxidants, the balance of which is obtained by redox regulation which involves redox sensing, signaling, responses, and feedback control pathways. This is part of our regulatory system and reduction of oxidative modification is fed by NADPH and NADH. Cellular protein thiols are the primary targets of redox modification and this is mediated by the GSH (thioredoxin and glutathione) systems (Sies et al., 2024) . When such restoration fails, a progressive state of allostatic load ensues. This load may suspend the proteins in a state of allostatic regulation—away from baseline conditions that support the participation of the proteins that are required for typical neurodevelopment and physical growth. More research needs to be done in this area as to which developmental proteins are disrupted via stress mechanisms, and how that impacts physical and neural development.
This realization became the foundation for what would ultimately evolve into my integrated framework: BioToggles, the biochemical network map, and the overarching model of Neurodivergent Biochemistry—a system in which redox shifts and protein isoform selection mediate systemic adaptations that manifest as both behavioral and physiological phenotypes. Existing in a state of allostasis vs baseline homeostasis.,
Biomarkers vs. Biochemical Pathways – Second Pattern Identified
Biochemistry stands apart from interpretive sciences because it is rooted in observable, reproducible processes governed by universal chemical and physical laws. Biochemical pathways (ranging from the TCA cycle and glucose metabolism to complex signaling cascades and neurotransmitter synthesis) operate with predictable precision, utilizing the same proteins and cofactors for each reaction across all humans.
Over 99.9% of protein-coding genes are conserved across individuals (Collins & Mansoura, 2001). However, due to genetic variation—including single nucleotide polymorphisms (SNPs), alternative splicing, and post-translational modifications , subtle differences can emerge in isoform expression or protein activity. Still, the core instructions for protein synthesis are encoded in DNA and can be directly measured and validated.
This inherent objectivity distinguishes biochemistry from fields that rely on statistical correlations or associative studies, which often depend on interpretive frameworks and are subject to revision as new data emerge. By utilizing metabolomics (Johnson et al., 2016), and comparing biochemical pathways with metabolite biomarker data in individuals who a given condition or diagnosis, researchers can establish a mechanistic understanding of how those pathways are functioning in those who have obtained that diagnosis—and how such disruptions influence the biochemical system as a whole. These comparisons enable precise identification of molecular dysfunctions underlying complex conditions, offering clarity where correlation-based approaches fall short. This becomes even more useful in individual analysis in what the physiological outcome of biochemical disruption would be for an individual.
Reverse Engineering via Biochemical Network Integration - Third Pattern Identified
The central dogma of molecular biology is often stated as: DNA → RNA → Protein (Mercadante et al., 2023). Through the construction of a biochemical network, I extended this sequence to include: Function within the cell → Function within the system, which allows for contextual analysis of how molecular-level changes cascade into system-wide effects.
A reverse engineering approach was employed to systematically deconstruct human physiology into its smallest functional units proteins (LaPelusa & Kashik, 2022) and to map their dynamic interactions within a comprehensive biochemical framework. Each gene encodes instructions for the synthesis of a protein and its isoforms. Protein variation is flexible and context-sensitive, enabling functional adaptation in response to environmental inputs and cellular stress. These proteins, acting as specialized molecular agents, execute tasks essential for maintaining cellular and systemic homeostasis. This context-sensitive variation in protein function is largely mediated by alternative splicing, an epigenetically regulated mechanism that expands proteomic diversity in response to developmental cues and cellular stressors (Luco et al., 2012).
To build the foundational network, curated data from the UniProt database were extracted and integrated. This included proprietary categorization methods developed to identify key regulatory dynamics. These elements collectively enabled the construction of a system-wide map of biochemical regulation.
This integrative approach enabled the identification of conserved yet stress-responsive proteins functioning as biochemical toggles—now defined in this framework as BioToggles. These toggles trigger the activation of specific protein isoforms in an effort to restore baseline homeostasis. When dysregulated under chronic stress, they contribute to systemic allostatic overload. The resulting network now functions as both a framework and a hypothesis-generating model, facilitating mechanistic inference from condition/diagnosis biomarker profiles.
Note: This model is not intended for clinical diagnostic use. It is designed exclusively for research and hypothesis generation, supporting the identification of biochemical patterns through cross-referencing biomarker data with known pathway dynamics.
Hypothesis Overview
Autism and its associated comorbidities are hypothesized to result from the chronic activation of allostatic pathways, initiated by epigenetic modifications, genetic mutations, or a combination of both. This hypothesis acknowledges the multifactorial nature of autism and the complex interplay of biological variables that shape its phenotypic expression (see The BH4 Pathway as an Allostatic Mechanism in the Pathology of Autism and Systemic Comorbidities Paper HERE)..
Within this framework, genetically or epigenetically triggered stress leaves the body in a prolonged state of allostasis, a dynamic effort to restore homeostasis under chronic cellular stress. During development, allostatically induced shifts in protein isoform expression and alterations in redox-sensitive proteins may be prioritized over normative developmental programs. This redirection of biochemical resources can impair neurodevelopment during critical windows, disrupt neurotransmitter dynamics within the Cortico-Striatal-Thalamic Loop (CSTL), and drive widespread oxidative stress. Over time, unresolved stress may result in allostatic overload, contributing to systemic comorbidities, chronic illness, and accelerated onset of age-related conditions such as neurodegenerative and neuromuscular disorders (see the Genomic and Proteomic Regulation in Cellular Homeostasis: From Molecular Mechanisms to Clinical Implications Paper HERE).
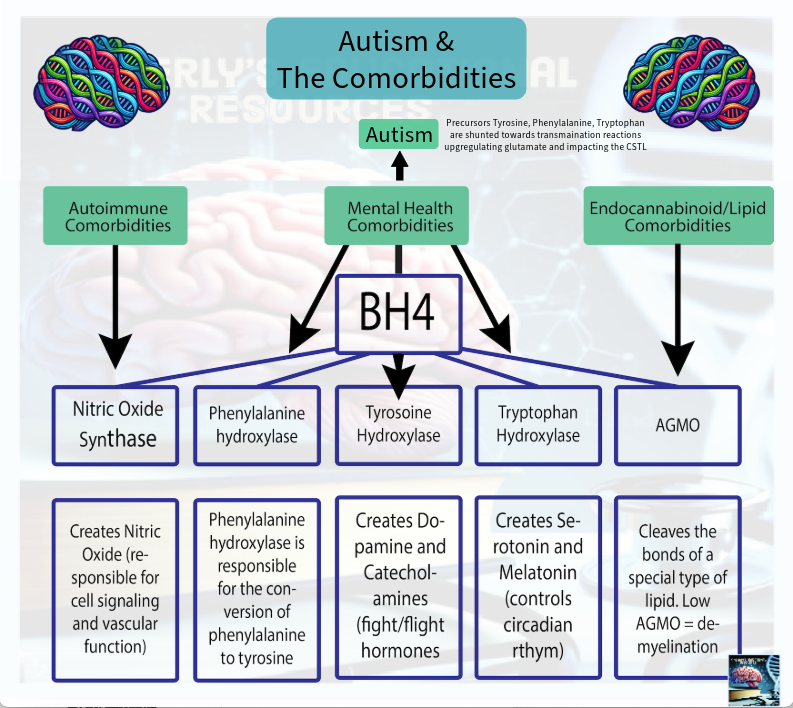
This hypothesis further proposes that the BH4 pathway functions not only as a cofactor system, but as an allostatic mechanism—a central biochemical lynchpin that responds dynamically to cellular redox shifts. Under cellular stress, BH4 is rerouted through a process defined here as the BH4 Shunt, which disrupts five key physiological domains both directly—via BH4-dependent enzymes such as NOS, AAAHs, and AGMO—and indirectly, through NOS uncoupling-induced redox shifts that activate a class of regulatory systems termed Epigenetic BioToggles. (see the Autism & the Comorbidities Along the BH4 Pathway Paper HERE).
These BioToggles coordinate adaptive changes across five interdependent domains: the immune system, metabolic pathways, nervous system, cellular repair mechanisms, and genomic/epigenetic protein regulation. While each toggle can be activated independently, their chronic and overlapping activation results in coordinated engagement of allostatic proteins across systems. These adaptive responses are designed to ensure survival during stress, but when persistently activated, they may ultimately compromise development and long-term physiological resilience. During allostatic states, the body may epigenetically reprioritize protein synthesis, favoring proteins essential for survival and stress resolution over those required for normative development. This shift in resource allocation may impact the availability and expression of proteins essential to neurodevelopment and systemic maturation during critical developmental windows. Further research is needed to fully understand how this stress-driven reprioritization alters long-term outcomes in individuals with autism phenotypes.
The specific phenotype that emerges—whether behavioral, physiological, or both—is determined by the initial BioToggle activation and the cascading downstream effects it exerts across the body’s interconnected biochemical systems.
Who Am I and Why Did I Do This?
I am an educator and neurodivergence advocate who collaborates with researchers and has developed a biochemical framework linking autism traits with their comorbid conditions. Inspired by my daughter’s nonverbal autism, I created NeuroToggle, a neuroplasticity approach that supported her transition to speech. You can read our story here. Building on this, I constructed the Autism and the Comorbidities Theory, which integrates autism biomarker data and proposes that genetically induced allostasis activates epigenetic effectors, including redox-sensitive protein shunts, with the BH4 Shunt as the central linchpin driving clusters of autism traits and comorbidities. To make this theory accessible, I created explanatory frameworks: NeuroToggle, the BioToggles, the BioDials, and Neurodivergent Biochemistry. I have disseminated this work through books, ResearchGate papers, websites, and social media. My work is timestamped and publicly documented, and I have reported instances of uncited overlap through appropriate research integrity channels.
You can find a full breakdown of the timeline here, with hyperlinks of evidence. You can also read my “about me” here.
Developing the Autism and the Comorbidities Theory
I released our story online. People asked if I had figured out a cure to autism. I clarified that I helped her with her comorbidity of being nonverbal, and that was purely anecdotal without further evidence. They asked what the comorbidities are and how they are linked to autism. This set me down the path of digging through autism research and creating a biochemical network to compare biomarker data to determine the biochemical link that link the physiological comorbidities to autism, first starting with fetal hyperglycemia. I documented the building of my biochemical network on TikTok through a living documentary.
In September 2023, I published Discovering Autism and the Comorbidities Along the BH4 Pathway, refining it into a theory that I call the Autism and the Comorbidities Theory (PHD Version and Laymen Version). In this theory, genetic mutations result in epigenetic redox protein shunts, with the BH4 Shunt as the center linchpin: NOS uncoupling activates other redox-sensitive shunts, and BH4 itself converts to BH2 under oxidative stress. These shifts produce classic checklist autism traits and major comorbidities (including autoimmune, mental health, and endocannabinoid dysregulation), while other redox-sensitive shunts account for additional physiological comorbidities, ultimately prioritizing stress responses over typical development and function.
Core claim. Genetic mutations result in allostasis induced regulated epigenetic redox protein shunts.
Central mechanisms. The BH4 Shunt acts as a center linchpin of our stress response, NOS uncoupling activates other redox-sensitive shunts, and BH4 itself converts to BH2 under oxidative stress. Genetic mutations that activate the regulatory system and BH4 Shunt results in classic checklist autism symptoms (from the BH4 Shunt induced transamination pathway upregulation dysregulating the E/I balance within the CSTL, oxytocin receptors in glutamatergic neurons in the PFC, as well as NOS uncoupling and AGMO related endocannabinoid system impairment.) with simultaneous comorbidities due to other epigenetic redox-sensitive protein shunts.
Consequence. These shifts prioritize the stress response over typical development and function, with allostatic overload leading to wear and tear over time.
*After forming my Autism and the Comorbidities Theory, it became clear that the VEGFA-NRP1 Pathway that controls facial motor neuron axon guidance to the facial muscles during fetal development is likely the epigenetic redox-sensitive protein shunt involved in nonverbality as a comorbidity. This may explain why neuroplasticity to motor neurons in my daughter’s facial muscles during critical time windows was productive in helping her acquire the ability to produce speech sounds that she previously did not have. It is mechanistically probable. However, that would require further research to confirm.
Frameworks to Explain and Apply the Theory and Concepts
To make the theory teachable and practically useful, I created educational and analytical frameworks for visual and explanatory purposes::
NeuroToggle — neuroplasticity strategies for building, expanding, strengthening, and timing neural connections (used in my daughter’s speech progress).
BioToggles — the five regulatory systems (immune, metabolic, cellular repair, nervous system, genetic regulation).
BioDials — time-based protein synthesis cycles (circadian, circannual, and developmental).
Neurodivergent Biochemistry — a systems-level integration explaining upstream and downstream impacts of allostasis in various contexts considering the variables of the BioToggles becoming situationally flipped, chronically stuck, or genetically locked and how that impacts the timer regulated flow-state of protein synthesis within the BioDials.
Together, these frameworks make the Autism and the Comorbidities Theory and Neurodivergent Biochemistry concepts accessible
The Redox Protein Shifts & The BH4 Shunt
The kinetics of biochemical pathways are entrained to the circadian rhythm under baseline conditions but may be disrupted during allostasis, as the body prioritizes the restoration of homeostasis in response to stress. These pathways are not autonomous—they require kinetic initiation and are reactive by nature. A stimulus is required to initiate any change in pathway activity. Under cellular stress the body toggles between two primary survival modes: repair and resilience to rest and digest at night (catabolic), or adaptation through resource storage to utilize at night and immune activation to protect ourselves in the day (anabolic). The BH4 Shunt model reveals how this bifurcation is biochemically governed by redox-sensitive mechanisms—allocating BH4 between cofactor-dependent repair processes and ROS-mediated catabolic signaling pathways. BH4 acts as the redox-sensitive biochemical lynchpin within this model, influencing each toggle: supporting or diverging nitric oxide synthase (NOS) in the immune system toggle; redirecting aromatic amino acid hydroxylases (AAAHs) in the nervous system toggle toward or away from dopamine, serotonin, and melatonin synthesis—shunting their precursor aromatic amino acids into transamination pathways; supporting alkylglycerol monooxygenase (AGMO) in the cellular repair toggle, as it is the only enzyme capable of cleaving ether lipid bonds; and contributing to metabolic adaptation via NOS uncoupling, which leads to reactive oxygen species (ROS) production. (Fanet et al., 2021).
Anabolic Mode: Rest and Digest (BH4-Depleted)
In the anabolic state, BH4 availability is preserved and directed toward its canonical cofactor functions. This upregulation is essential for BH4's function as an anti-inflammatory molecule and free radical scavenger, helping to control oxidative stress and maintain immune balance. This supports system-wide biochemical stability as we rest:
NOS Pathway (Immune Toggle): BH4 enables coupled nitric oxide synthesis via NOS enzymes, producing NO instead of superoxide. This supports vascular regulation, pathogen defense, and immune resolution, particularly during short-term autoinflammatory responses.
AAAH Pathway (Nervous System Toggle): BH4 serves as a cofactor for aromatic amino acid hydroxylases(tyrosine hydroxylase, tryptophan hydroxylase), enabling the synthesis of dopamine, serotonin, and melatonin. These neurotransmitters regulate mood, sleep, sensory processing, and stress recovery.
AGMO Pathway (Cellular Repair Toggle): BH4 supports alkylglycerol monooxygenase, the only enzyme that cleaves ether lipids. This facilitates membrane remodeling, detoxification of lipid peroxidation byproducts, and lysosomal maintenance, protecting against cellular debris accumulation.
mTORC1 Regulation (Metabolic Toggle): Adequate BH4 function indirectly stabilizes mTORC1 signaling by maintaining lysosomal integrity and redox balance, allowing for proper nutrient sensing, autophagy cycling, and balanced energy utilization.
Catabolic Mode: Activation and Storage (BH4-Supported)
When a situationally induced BioToggle results in oxidative stress, or when genetically induced BioToggle activation shifts baseline priorities, BH4 is oxidized to BH2 and diverted from its repair functions. This initiates a catabolic program characterized by resource mobilization and inflammatory adaptation:
NOS Uncoupling (Immune Toggle): In chronic immune activation—especially in autoimmune responses—NOS becomes uncoupled due to BH4 depletion. This produces superoxide instead of nitric oxide, escalating ROS levels and driving a self-sustaining inflammatory loop (Fanet et al., 2021).
Neurotransmitter Deficiency (Nervous System Toggle): AAAH activity is impaired, reducing dopamine, serotonin, and melatonin. Their precursor amino acids are instead shunted into transamination pathways, increasing glutamate production, disrupting excitatory/inhibitory balance, and contributing to neurobehavioral symptoms and sensory dysregulation.
AGMO Inhibition and Lysosomal Stress (Cellular Repair Toggle): Without BH4, AGMO function declines, resulting in ether lipid accumulation and lysosomal dysfunction. Impaired autophagy and defective membrane turnover contribute to cellular overload and reduced detoxification capacity.
mTORC1 Disruption (Metabolic Toggle): Amino acid depletion from transamination up regulation, and lysosomal stress from ROS accumulation lead to mTORC1 being inhibited, leading to the dephosphorylation and nuclear translocation of TFEB and TFE3. Once in the nucleus, these transcription factors activate genes involved in lysosomal biogenesis, autophagy, and metabolic stress adaptation. The resulting proteins represent a coordinated allostatic response designed to restore cellular homeostasis and resolve accumulated stress signals. (Uniprot 42345).
Side note: mTOR has been implicated in prior research in aberrant pruning during the two developmental periods in ASD (Faust et al., 2021).
Epigenetic Reprogramming (Genomic Toggle): Redox-sensitive transcription factors (ex: NF-κB, NRF2, HIF-1α) are activated, favoring stress-response gene expression over developmental programs. This leads to persistent BioToggle engagement and long-term isoform selection shifts, further embedding maladaptive biochemical states, leading to allostatic overload.
BH4 is also a responder to changes in state of each of the BioToggles:
Immune toggle: Inflammation and oxidative stress oxidize BH4 to BH2.
Metabolic toggle: Alters BH4 regeneration via the folate pathway (DHFR).
Nervous system toggle: Increases BH4 demand for AAAH-dependent neurotransmitter synthesis.
Cellular repair toggle: Requires BH4 for AGMO activity during lipid remodeling.
Genomic regulation toggle: Adjusts transcription of BH4-dependent enzymes under stress-responsive control.
Building upon the established concepts of allostatic load and allostatic overload, I introduced the complementary framework of systemic load and systemic overload to describe how redox shifts function as early cellular warning signals that ultimately initiate allostatic responses. These shifts precede full allostatic load and overload, and represent the regulatory system's attempt to rebalance under duress. In the context of autism, these compensatory adaptations may deprioritize neurodevelopmental processes in favor of restoring baseline homeostasis, particularly during genetically induced allostasis resulting from mutations impacting the nervous system BioToggles. This disruption can impair the development of key neural pathways involved in communication, cognition, and regulatory function during critical developmental windows. Comorbidities may also emerge due to the concurrent strain placed on other toggles, their progression toward allostatic overload, and the dysregulation of BH4-dependent pathways during both development and aging.
The BH4 Shunt is not merely a passive redistribution of a cofactor—it is a central decision point in cellular biochemistry that determines whether the body pursues repair or adaptation. When BH4 is preserved and allocated toward its developmental and homeostatic functions, the system fosters resilience and recovery. When BH4 is chronically/permanantly shunted—whether through situational or genetically induced BioToggle activation—the system enters a chronic anabolic/catabolic states, depending on the initiating stressor.
Chronic allostasis, driven by epigenetic and genetic factors, initiates systemic changes that begin early and compound over time—altering how the brain and body develop in childhood, function across the lifespan, and ultimately age:
Neurodevelopment: BioToggle-induced epigenetic variation in protein isoform expression, along with the activation of allostatic pathways, may redirect biochemical signaling during critical developmental windows. This shift appears to prioritize cellular restoration to baseline over the expression of developmental proteins, potentially altering neural structures and connections that shape autistic neurodevelopmental trajectories. This is functionally known, but requires more research into impact and impact on various phenotypes.
Immediately Noticeable Symptoms:
Symptomatic changes in neural circuitry—particularly within the Cortico-Striatal-Thalamic Loop (CSTL), a network critical for motor planning, cognitive flexibility, emotional regulation, and executive function. These CSTL-specific disruptions contribute directly to many of the core behavioral characteristics associated with autism, and has been heavily linked to autism in the existing peer reviewed literature (Soghomonian, 2024; Abbot et al., 2018; Li & Pozzo-miller, 2020; Fuccillo, 2016; Di Martino et al., 2011).
More research is needed into how immediate changes to the ECM due to chronic oxidative stress will impact hEDS due to the redox sensitive MMPs that degrade all the components of the extracellular matrix (Lu & Wahl, 2005),, and in POTS due to the impact of redox sensitive ACE2 (Rabelo et al., 2011; Uniprot Q9BYF1) on the RAS system (Fountain et al., 2023; Kitzerow, 2024). Both hEDS and POTS have shown to be prevalently comorbid in autistic individuals (Owens et al., 2021).
Redox sensitive disruption of facial motor neuron development may directly contribute to the atypical facial musculature often observed in autism and, in some cases, may affect the ability to produce speech. As previously discussed, facial motor neurons originate from rhombomere 4 and depend on tightly regulated axon guidance mechanisms—including the VEGFA–NRP1 signaling—for proper migration and synaptic targeting (Raimondi et al., 2016; Uniprot P15692; Uniprot 014786). These neurons innervate muscles responsible for facial expression, articulation, and oral-motor control. When guidance cues are disrupted or neuronal survival is compromised, the corresponding muscles may experience atrophy or remain hypotonic due to insufficient trophic support. This form of neurogenic hypotonia may underlie several features commonly reported in autism, including reduced oral-motor tone, inefficient articulation, flat affect, nonverbality, and incongruent or absent facial expressions (Grossard et al., 2020; Press et al., 2010; Brewer et al., 2016; Weiss et al., 2021; Drimalla et al., 2021; Maffei et al., 2024; Foster et al., 2024; Trevisan et al., 2018).
However, even with all this data indicating facial muscle oddities in autistics, not one physical inside scan of the motor nerves/muscles involved with the production of speech or facial expressions has been done to test for functionality of these mechanisms/parts. These deficits have only ever been considered as social not functional….
Decline Over Time: This bifurcation underlies the physiological divergence observed in autism, autoimmune conditions, metabolic syndromes, and neurodegenerative diseases, linking BH4 availability to systemic health outcomes across the lifespan. Over time, sustained allostatic activity may contribute to physiological wear and tear, which has been associated with an increased incidence of early-onset age-related conditions in autistic populations (Klein & Klinger, 2024; Mason et al., 2022).
Personal Catalyst: My “Why”
This work was born out of personal necessity. In October 2020, I noticed my daughter couldn’t blow out the candles on her birthday cake. That moment shifted my understanding of her nonverbal autism from a behavioral concern to a physiological one. My daughter was formally diagnosed with nonverbal autism in April 2021 and placed on long-term disability services through the Children’s Long-Term Support (CLTS) Program and Katie Beckett Medicaid. She began ABA therapy shortly after, but her behaviors worsened. The ABA framework does not consider behavior as a form of communication in the core functions of behavior for nonverbal kids, nor does it factor autonomic dysregulation for behaviors for autistics.
We had added oral motor exercises—starting with blowing out candles—and over time, I began developing a targeted neuroplasticity plan. I turned to academic journal databases to search for peer-reviewed research on enhancing neurodevelopment outcomes and realized that each skill or behavior could be understood as a neural circuit. With this in mind, I structured an intervention framework grounded in educational pedagogy, designed to guide these circuits using instructional strategies. By February 2022, this plan had evolved into the foundational structure for what would later become NeuroToggle.
In November 2022, I publicly shared her progress—at that point, she had gained some functional speech. By April 2024, she was fully verbal, socially engaged, and no longer functionally eligible for services. She still requires an IEP for reading and intervention support, but she has come a long way in the last few years. At this time she was disenrolled from CLTS following an evaluation and considered no longer functionally eligible for services.
By January 2024, I formally published my hypothesis on ResearchGate in the form of a peer-accessible white paper titled "Autism & the Comorbidities Along the BH4 Pathway," followed by two additional papers exploring the broader genomic and proteomic regulatory networks that influence BH4-dependent pathways and stress adaptation, as well as further biochemical insights into the BH4 pathway and related autism comorbidities.
In September 2024, I introduced the Allostatic Toggles Framework, categorizing physiological stress responses into toggle-based systems that dynamically regulate homeostasis across immune, metabolic, nervous system, cellular repair, and genomic domains. This framework evolved into the Epigenetic BioToggles, providing a model for understanding how environmental and internal stressors influence epigentic biochemical adaptation and how, when left on too long, may result in long-term health outcomes due to allostatic overload.
The term NeuroToggle was formally trademarked January 2025, and BioToggle was formally trademarked in April 2025 to protect the broader biochemical and educational framework associated with these discoveries.
📖 Core Definitions
My Contributions
Systemic Load: The total burden placed on the body by internal and external stressors across systems at a given moment.
Systemic Overload: A spike in total stress burden that threatens to trigger toggle activation or overwhelm regulatory capacity.
BH4 Shunt: The BH4 Shunt refers to the dynamic, redox-sensitive mechanism by which tetrahydrobiopterin (BH4) is both diverted from its canonical cofactor functions and reallocated across stress-response pathways during allostatic conditions. In the Neurodivergent Biochemistry framework, BH4 is positioned as a biochemical lynchpin that plays a bidirectional role: it both regulates and responds to biochemical changes within each of the five BioToggles—immune, metabolic, nervous system, cellular repair, and genomic regulation.
As a Responder - BH4 levels, redox state, and utilization patterns shift in response to stress-induced activation of BioToggles:
Immune toggle: Inflammation and oxidative stress oxidize BH4 to BH2.
Metabolic toggle: Alters BH4 regeneration via the folate pathway.
Nervous system toggle: Increases BH4 demand for AAAH-dependent neurotransmitter synthesis.
Cellular repair toggle: Requires BH4 for AGMO activity during lipid remodeling.
Genomic regulation toggle: Adjusts transcription of BH4-dependent enzymes under stress-responsive control.
As a Regulator - The redistribution and redox modulation of BH4 directly influence toggle behavior:
NOS uncoupling (due to BH4 loss) drives ROS production, sustaining immune and metabolic toggle engagement.
AAAH substrate shunting affects dopamine, serotonin, and melatonin availability, reshaping nervous system signaling.
AGMO inhibition impairs ether lipid cleavage, compromising membrane repair and detoxification.
mTOR and redox-sensitive protein signaling respond to ROS increases stemming from BH4 oxidation, recalibrating growth, metabolism, and repair.
This dual role of BH4 reflects a tightly coupled feedback circuit in which it acts both as a sensor of redox state and as a driver of adaptive biochemical prioritization. Under prolonged allostatic conditions, BH4 shunting may result in the epigenetic reprioritization of protein isoform expression—favoring immediate survival and stress-resolution processes over normative developmental functions. This shift has profound implications for neurodevelopmental timing, cognitive outcomes, and the emergence of systemic comorbidities.
Neurodivergent Biochemistry refers to the study of how biochemical processes adapt under chronic stress to produce neurodevelopmental divergence and systemic comorbidities. It emphasizes the role of redox-sensitive feedback, protein isoform selection, and allostatic prioritization in shaping both behavior and physiology. Within this framework, stress-induced disruptions in biochemical homeostasis—mediated by the activation of Epigenetic BioToggles—alter systemic regulation in ways that deviate from typical developmental trajectories. Neurodivergent Biochemistry provides a mechanistic lens for understanding how biochemical shifts driven by environmental and genetic stressors contribute to autism and related conditions.
Existing Definitions
Allostasis: The body's dynamic, adaptive response to stress, using biochemical shifts to maintain stability through change.
Allostatic Load: The cumulative wear on systems from repeated or prolonged activation of allostasis in response to stress.
Allostatic Overload: A state in which the body’s adaptive capacity is exceeded, leading to system wear and tear that may lead to health implications.
📚 Primary Lit Reviews:
All titles authored by Kimberly Kitzerow and published on ResearchGate:
The BH4 Pathway as an Allostatic Mechanism in the Pathology of Autism and Systemic Comorbidities
August 2024 | DOI: 10.13140/RG.2.2.19905.98401/2
August 2024 | DOI: 10.13140/RG.2.2.31853.19682/3
Autism & the Comorbidities Along the BH4 Pathway
January 2024 |. DOI: 10.13140/RG.2.2.23124.37761
📘 Applied Neuroplasticity Publication
NeuroToggle
(Amazon KDP, October 2024)
This companion work outlines the applied neuroplasticity framework I developed as an educational model to support my daughter's transition from nonverbal autism to speech. It complements the biochemical model by demonstrating targeted instructional strategies for building neural circuits in practice. This framework is rooted exclusively in educational pedagogy and is designed for instructional use. It is not intended to function as a medical intervention, therapeutic protocol, or clinical treatment.
Trademark registered January 11, 2025.
📅 Timeline of Disclosure
October 2020: Recognized oral-motor deficits when daughter couldn’t blow out birthday candles.
April 2021: Daughter diagnosed with nonverbal autism;
Shortly After, but unknown exact date (I was in survival mode): daughter placed on CLTS and Katie Beckett and began ABA.
February 2022: Began structured neuroplasticity plan (foundation of NeuroToggle), stopped ABA by summer.
November 2022: Shared public progress—functional speech achieved. When asked how Kylie's nonverbality related to her autism, I began mapping out the connection visually using Adobe Illustrator, arranging published data into a jigsaw puzzle format to search for overlapping commonalities. It became clear to me that the comorbidities and autism traits observed in the same subset of individuals likely shared a simultaneous root cause. This led me to discover converging patterns in the BH4 pathway, linking autism biomarkers and comorbidities through what I would later define as the BH4 shunt. I realized that these patterns centered on redox-sensitive proteins—each impacted in service of shifting the body's priorities into allostatic adaptation vs baseline conditions that support typical neurodevelopment and physical development.
May 2023: Initial hypothesis shared via TikTok video. Continued to document new BH4 pathway links and development of my hypothesis in my “Autism Pathology Journey” Playlist on TikTok.
January 2024: First formal publication (ResearchGate).
April 2024: Disenrolled from disability services after being deemed no longer functionally eligible.
August 2024: Expanded biochemical and genomic frameworks released.
August 2024: Allostatic Toggles Famework released.
October 2024: NeuroToggle published on Amazon.
January 2025: NeuroToggle trademark registered.
April 2025: BioToggle trademark registered.
🔐 Attribution & Citation Notice:
This document formally timestamps the original discovery and framework development of the BioToggle hypothesis, the Epigenetic BioToggles system, and related publications by Kimberly Kitzerow. Any derivative use, academic citation, or conceptual adaptation must credit Kimberly Kitzerow as the originator of the hypothesis and framework.
For more information, citations, and visual frameworks: www.kimberlyedu.org
For questions, collaborations, or verification please email: kitzerow@kimberlyedu.org
References:
Hoffman KW, Tran KT, Moore TM, Gataviņš MM, Visoki E, DiDomenico GE, Schultz LM, Almasy L, Hayes MR, Daskalakis NP, Barzilay R. Allostatic load in early adolescence: gene / environment contributions and relevance for mental health. medRxiv [Preprint]. 2023 Oct 28:2023.10.27.23297674. doi: 10.1101/2023.10.27.23297674. PMID: 37961462; PMCID: PMC10635214.
McEwen BS, Wingfield JC. The concept of allostasis in biology and biomedicine. Horm Behav. 2003 Jan;43(1):2-15. doi: 10.1016/s0018-506x(02)00024-7. PMID: 12614627.
Guidi J, Lucente M, Sonino N, Fava GA. Allostatic Load and Its Impact on Health: A Systematic Review. Psychother Psychosom. 2021;90(1):11-27. doi: 10.1159/000510696. Epub 2020 Aug 14. PMID: 32799204.
McEwen BS, Gianaros PJ. Stress- and allostasis-induced brain plasticity. Annu Rev Med. 2011;62:431-45. doi: 10.1146/annurev-med-052209-100430. PMID: 20707675; PMCID: PMC4251716.
Blair C, Raver CC, Granger D, Mills-Koonce R, Hibel L; Family Life Project Key Investigators. Allostasis and allostatic load in the context of poverty in early childhood. Dev Psychopathol. 2011 Aug;23(3):845-57. doi: 10.1017/S0954579411000344. PMID: 21756436; PMCID: PMC4167021.
Danese A, McEwen BS. Adverse childhood experiences, allostasis, allostatic load, and age-related disease. Physiol Behav. 2012 Apr 12;106(1):29-39. doi: 10.1016/j.physbeh.2011.08.019. Epub 2011 Aug 25. PMID: 21888923.
Kallen V, Tahir M, Bedard A, Bongers B, van Riel N, van Meeteren N. Aging and Allostasis: Using Bayesian Network Analytics to Explore and Evaluate Allostatic Markers in the Context of Aging. Diagnostics (Basel). 2021 Jan 21;11(2):157. doi: 10.3390/diagnostics11020157. PMID: 33494482; PMCID: PMC7912325.
Arnsten AF. Stress signalling pathways that impair prefrontal cortex structure and function. Nat Rev Neurosci. 2009 Jun;10(6):410-22. doi: 10.1038/nrn2648. PMID: 19455173; PMCID: PMC2907136.
Mualem R, Morales-Quezada L, Farraj RH, Shance S, Bernshtein DH, Cohen S, Mualem L, Salem N, Yehuda RR, Zbedat Y, Waksman I, Biswas S. Econeurobiology and brain development in children: key factors affecting development, behavioral outcomes, and school interventions. Front Public Health. 2024 Sep 26;12:1376075. doi: 10.3389/fpubh.2024.1376075. PMID: 39391155; PMCID: PMC11465878.
Martel AC, Galvan A. Connectivity of the corticostriatal and thalamostriatal systems in normal and parkinsonian states: An update. Neurobiol Dis. 2022 Nov;174:105878. doi: 10.1016/j.nbd.2022.105878. Epub 2022 Sep 29. Erratum in: Neurobiol Dis. 2023 Feb;177:105985. doi: 10.1016/j.nbd.2022.105985. PMID: 36183947; PMCID: PMC9976706.
Al-Beltagi M. Autism medical comorbidities. World J Clin Pediatr. 2021 May 9;10(3):15-28. doi: 10.5409/wjcp.v10.i3.15. PMID: 33972922; PMCID: PMC8085719.
Chung US, Kim JH. Common Comorbid Condition of Patients With Autism Spectrum Disorder and Pharmacotherapy for Patients With Autism Spectrum Disorder. J Korean Acad Child Adolesc Psychiatry. 2024 Jan 1;35(1):39-43. doi: 10.5765/jkacap.230006. PMID: 38204750; PMCID: PMC10774559.
Khachadourian V, Mahjani B, Sandin S, Kolevzon A, Buxbaum JD, Reichenberg A, Janecka M. Comorbidities in autism spectrum disorder and their etiologies. Transl Psychiatry. 2023 Feb 25;13(1):71. doi: 10.1038/s41398-023-02374-w. PMID: 36841830; PMCID: PMC9958310.
Bougeard C, Picarel-Blanchot F, Schmid R, Campbell R, Buitelaar J. Prevalence of Autism Spectrum Disorder and Co-morbidities in Children and Adolescents: A Systematic Literature Review. Front Psychiatry. 2021 Oct 27;12:744709. doi: 10.3389/fpsyt.2021.744709. PMID: 34777048; PMCID: PMC8579007.
Burns J, Phung R, McNeill S, Hanlon-Dearman A, Ricci MF. Comorbidities Affecting Children with Autism Spectrum Disorder: A Retrospective Chart Review. Children (Basel). 2023 Aug 19;10(8):1414. doi: 10.3390/children10081414. PMID: 37628413; PMCID: PMC10453739.
Hours C, Recasens C, Baleyte JM. ASD and ADHD Comorbidity: What Are We Talking About? Front Psychiatry. 2022 Feb 28;13:837424. doi: 10.3389/fpsyt.2022.837424. PMID: 35295773; PMCID: PMC8918663.
Sies H, Mailloux RJ, Jakob U. Fundamentals of redox regulation in biology. Nat Rev Mol Cell Biol. 2024 Sep;25(9):701-719. doi: 10.1038/s41580-024-00730-2. Epub 2024 Apr 30. Erratum in: Nat Rev Mol Cell Biol. 2024 Sep;25(9):758. doi: 10.1038/s41580-024-00754-8. PMID: 38689066; PMCID: PMC11921270.
Collins FS, Mansoura MK. The Human Genome Project. Revealing the shared inheritance of all humankind. Cancer. 2001 Jan 1;91(1 Suppl):221-5. doi: 10.1002/1097-0142(20010101)91:1+<221::aid-cncr8>3.3.co;2-0. PMID: 11148583.
Johnson CH, Ivanisevic J, Siuzdak G. Metabolomics: beyond biomarkers and towards mechanisms. Nat Rev Mol Cell Biol. 2016 Jul;17(7):451-9. doi: 10.1038/nrm.2016.25. Epub 2016 Mar 16. PMID: 26979502; PMCID: PMC5729912.
Mercadante AA, Dimri M, Mohiuddin SS. Biochemistry, Replication and Transcription. [Updated 2023 Aug 14]. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2025 Jan-. Available from: https://www.ncbi.nlm.nih.gov/books/NBK540152/
LaPelusa A, Kaushik R. Physiology, Proteins. [Updated 2022 Nov 14]. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2025 Jan-. Available from: https://www.ncbi.nlm.nih.gov/books/NBK555990/
Luco RF, Allo M, Schor IE, Kornblihtt AR, Misteli T. Epigenetics in alternative pre-mRNA splicing. Cell. 2011 Jan 7;144(1):16-26. doi: 10.1016/j.cell.2010.11.056. PMID: 21215366; PMCID: PMC3038581.
Fanet H, Capuron L, Castanon N, Calon F, Vancassel S. Tetrahydrobioterin (BH4) Pathway: From Metabolism to Neuropsychiatry. Curr Neuropharmacol. 2021;19(5):591-609. doi: 10.2174/1570159X18666200729103529. PMID: 32744952; PMCID: PMC8573752.
Faust TE, Gunner G, Schafer DP. Mechanisms governing activity-dependent synaptic pruning in the developing mammalian CNS. Nat Rev Neurosci. 2021 Nov;22(11):657-673. doi: 10.1038/s41583-021-00507-y. Epub 2021 Sep 20. PMID: 34545240; PMCID: PMC8541743.
Soghomonian JJ. The cortico-striatal circuitry in autism-spectrum disorders: a balancing act. Front Cell Neurosci. 2024 Jan 11;17:1329095. doi: 10.3389/fncel.2023.1329095. PMID: 38273975; PMCID: PMC10808402.
Abbott AE, Linke AC, Nair A, Jahedi A, Alba LA, Keown CL, Fishman I, Müller RA. Repetitive behaviors in autism are linked to imbalance of corticostriatal connectivity: a functional connectivity MRI study. Soc Cogn Affect Neurosci. 2018 Jan 1;13(1):32-42. doi: 10.1093/scan/nsx129. PMID: 29177509; PMCID: PMC5793718.
Li W, Pozzo-Miller L. Dysfunction of the corticostriatal pathway in autism spectrum disorders. J Neurosci Res. 2020 Nov;98(11):2130-2147. doi: 10.1002/jnr.24560. Epub 2019 Nov 22. PMID: 31758607; PMCID: PMC7242149.
Fuccillo MV. Striatal Circuits as a Common Node for Autism Pathophysiology. Front Neurosci. 2016 Feb 9;10:27. doi: 10.3389/fnins.2016.00027. PMID: 26903795; PMCID: PMC4746330.
Di Martino A, Kelly C, Grzadzinski R, Zuo XN, Mennes M, Mairena MA, Lord C, Castellanos FX, Milham MP. Aberrant striatal functional connectivity in children with autism. Biol Psychiatry. 2011 May 1;69(9):847-56. doi: 10.1016/j.biopsych.2010.10.029. Epub 2010 Dec 31. PMID: 21195388; PMCID: PMC3091619.
Lu Y, Wahl LM. Oxidative stress augments the production of matrix metalloproteinase-1, cyclooxygenase-2, and prostaglandin E2 through enhancement of NF-kappa B activity in lipopolysaccharide-activated human primary monocytes. J Immunol. 2005 Oct 15;175(8):5423-9. doi: 10.4049/jimmunol.175.8.5423. PMID: 16210649.
Rabelo LA, Alenina N, Bader M. ACE2-angiotensin-(1-7)-Mas axis and oxidative stress in cardiovascular disease. Hypertens Res. 2011 Feb;34(2):154-60. doi: 10.1038/hr.2010.235. Epub 2010 Dec 2. PMID: 21124322.
Fountain JH, Kaur J, Lappin SL. Physiology, Renin Angiotensin System. [Updated 2023 Mar 12]. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2025 Jan-. Available from: https://www.ncbi.nlm.nih.gov/books/NBK470410/
Owens AP, Mathias CJ, Iodice V. Autonomic Dysfunction in Autism Spectrum Disorder. Front Integr Neurosci. 2021 Dec 30;15:787037. doi: 10.3389/fnint.2021.787037. PMID: 35035353; PMCID: PMC8756818.
Raimondi C, Brash JT, Fantin A, Ruhrberg C. NRP1 function and targeting in neurovascular development and eye disease. Prog Retin Eye Res. 2016 May;52:64-83. doi: 10.1016/j.preteyeres.2016.02.003. Epub 2016 Feb 27. PMID: 26923176; PMCID: PMC4854174.
Grossard C, Dapogny A, Cohen D, Bernheim S, Juillet E, Hamel F, Hun S, Bourgeois J, Pellerin H, Serret S, Bailly K, Chaby L. Children with autism spectrum disorder produce more ambiguous and less socially meaningful facial expressions: an experimental study using random forest classifiers. Mol Autism. 2020 Jan 13;11(1):5. doi: 10.1186/s13229-020-0312-2. PMID: 31956394; PMCID: PMC6958757.
Press C, Richardson D, Bird G. Intact imitation of emotional facial actions in autism spectrum conditions. Neuropsychologia. 2010 Sep;48(11):3291-7. doi: 10.1016/j.neuropsychologia.2010.07.012. Epub 2010 Jul 16. PMID: 20638398; PMCID: PMC3221037.
Brewer R, Biotti F, Catmur C, Press C, Happé F, Cook R, Bird G. Can Neurotypical Individuals Read Autistic Facial Expressions? Atypical Production of Emotional Facial Expressions in Autism Spectrum Disorders. Autism Res. 2016 Feb;9(2):262-71. doi: 10.1002/aur.1508. Epub 2015 Jun 6. PMID: 26053037; PMCID: PMC4975602.
Weiss EM, Rominger C, Hofer E, Fink A, Papousek I. Less differentiated facial responses to naturalistic films of another person's emotional expressions in adolescents and adults with High-Functioning Autism Spectrum Disorder. Prog Neuropsychopharmacol Biol Psychiatry. 2019 Mar 8;89:341-346. doi: 10.1016/j.pnpbp.2018.10.007. Epub 2018 Oct 16. PMID: 30336172.
Drimalla H, Baskow I, Behnia B, Roepke S, Dziobek I. Imitation and recognition of facial emotions in autism: a computer vision approach. Mol Autism. 2021 Apr 6;12(1):27. doi: 10.1186/s13229-021-00430-0. PMID: 33823922; PMCID: PMC8025560.
Maffei MF, Chenausky KV, Haenssler A, Abbiati C, Tager-Flusberg H, Green JR. Exploring Motor Speech Disorders in Low and Minimally Verbal Autistic Individuals: An Auditory-Perceptual Analysis. Am J Speech Lang Pathol. 2024 May;33(3):1485-1503. doi: 10.1044/2024_AJSLP-23-00237. Epub 2024 Mar 21. PMID: 38512040.
Klein CB, Klinger LG. Aging Well and Autism: A Narrative Review and Recommendations for Future Research. Healthcare (Basel). 2024 Jun 17;12(12):1207. doi: 10.3390/healthcare12121207. PMID: 38921321; PMCID: PMC11203987.
Mason D, Ronald A, Ambler A, Caspi A, Houts R, Poulton R, Ramrakha S, Wertz J, Moffitt TE, Happé F. Autistic traits are associated with faster pace of aging: Evidence from the Dunedin study at age 45. Autism Res. 2021 Aug;14(8):1684-1694. doi: 10.1002/aur.2534. Epub 2021 May 27. PMID: 34042279; PMCID: PMC8328948.
González-Herrero B, Morgante F, Pagonabarraga J, Stanton B, Edwards MJ. Autism Spectrum Disorder May Be Highly Prevalent in People with Functional Neurological Disorders. J Clin Med. 2022 Dec 30;12(1):299. doi: 10.3390/jcm12010299. PMID: 36615098; PMCID: PMC9821674.